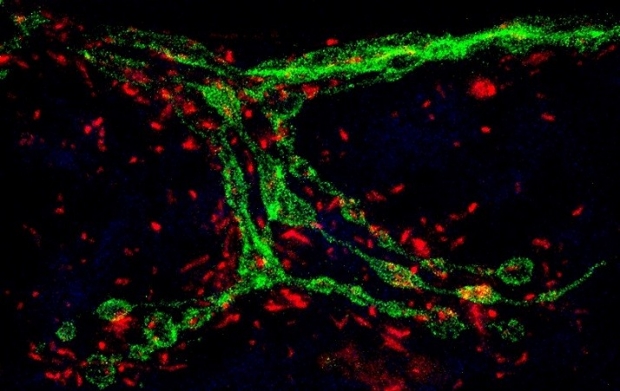
Our research lies at the intersection of mitochondrial cell biology and the mechanisms of neurological disorders, because of the vital importance of mitochondria to neuronal function and neuropathology. The dynamic network of mitochondria undergoes structural and functional adaptations in response to cell-type-specific metabolic demands. The multifaceted roles of mitochondria for ATP supply, Ca++ homeostasis and apoptosis support unique subcellular functions and are especially crucial to neurons, because the complex shape of a neuron poses spatiotemporal metabolic burden. In addition, defective mitochondria, which can be highly deleterious to a cell because of their output of reactive oxygen species (ROS), need to be repaired by fusing with healthy mitochondria or cleared from the cell. Mitochondria are also the information processing center of the cell. They are a reservoir of metabolites, small molecules, ions, ROS which can all function as signaling messengers to communicate with and influence other organelles and cellular processes. Thus, mitochondrial cell biology poses critical questions for all cells, but especially for neurons: how the cell sets up an adequate distribution of the organelle; how it sustains mitochondria in the periphery; how mitochondria are removed after damage; and how mitochondria communicate with other organelles. The goal of our research is to understand the central role of mitochondria in neuronal energy homeostasis, communications, and signal transduction, which provides a framework to probe their significance in health and disease.
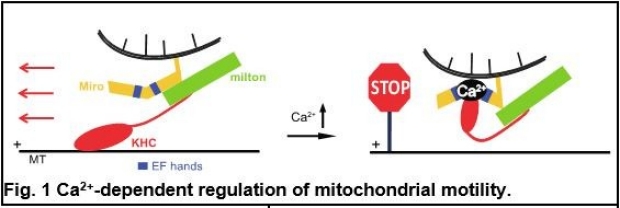
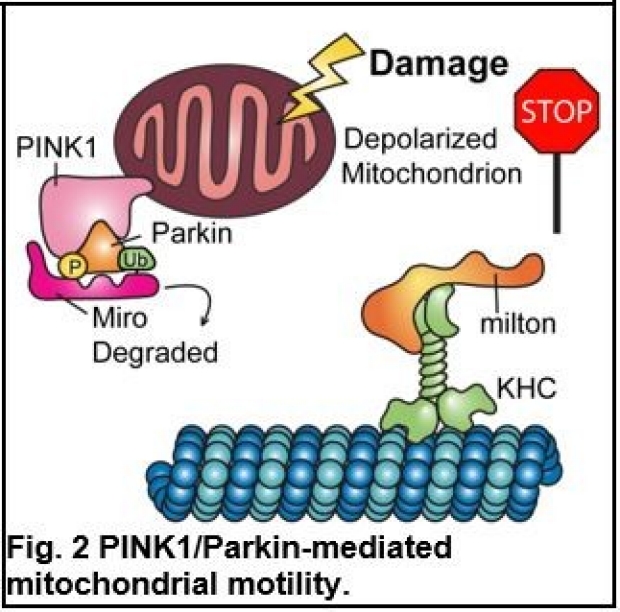
Our earlier research has provided fundamental insights into how different cellular signals regulate mitochondrial motility by controlling a motor/adaptor complex (Miro/milton/KHC) distinctively, in response to temporal and spatial needs. The sensitivity of mitochondrial movement to Ca2+ is likely to serve as a means by which mitochondria can be recruited to areas of high metabolic demand or low local ATP, and Ca2+ achieves this through changes in the conformation of the motor/adaptor complex transiently and instantaneously (Fig.1) (1). In contrast, when mitochondria are damaged, Miro in the motor/adaptor complex is subject to proteasomal degradation, which is primed by a Ser/Thr kinase PINK1 and the subsequent action of an E3 ubiquitin ligase Parkin, thereby arresting mitochondria (Fig. 2) (2). Stopping mitochondria in this manner is an early step in the quarantining of damaged mitochondria for subsequent degradation via mitophagy (Fig. 3). PINK1 and Parkin are two Parkinson’s disease (PD)-associated proteins. Based on this work, recently we have extended this basic understanding of mitochondrial motility and quality control to PD pathogenesis. We have found that in PD patients with mutations in PINK1 or Parkin, a failure to stop, isolate, and remove the damaged mitochondria likely contributes to neuronal cell death (3-5).
The importance of the mitochondrial outer membrane (OMM) protein Miro in mediating mitochondrial motility and safeguarding mitochondrial quality has led us to examine Miro in depth in multiple types of PD. We have found that Miro can interact with additional PD proteins, such as LRRK2 and alpha-synuclein. Intriguingly, Miro is accumulated on the damaged mitochondria in human neurons from both hereditary and sporadic PD patients, even without any known mutations, leading to slower mitophagy and oxidative stress (Fig.3) (3-5). This data suggests Miro-mediated mitophagy may represent a common denominator underlying different types of PD. To determine whether Miro is an effective target for PD, we have utilized the artificial intelligence technology that tackles complex drug discovery optimization, to search for small molecules from a vast chemical space in silico that are predicted to bind to Miro1. We have discovered a compound series that reduces Miro1, named Miro1 Reducers (3, 6). Treating fibroblasts from PD patients with the lead compound eliminates the Miro1 molecular pathology, and treating PD flies and patient-derived neurons with the compound rescues their locomotor deficits and dopaminergic neurodegeneration (3, 6). The small molecules discovered are predicted to be suitable for oral uptake, be free of toxicity, and pass the blood-brain barrier. Thus, those Miro1 Reducers could form a new class of compounds to combat PD in clinical trials. A major roadblock in current PD clinical trials is the lack of pharmacodynamic markers for target engagement assessment. To overcome this challenge, we have found that Miro1 is a reliable marker for classifying PD and monitoring the efficacy of Miro1 Reducers in 3 independent PD cohorts (3, 7, 8). Most recently, we have developed a blood test of Miro1 using a high throughput ELISA assay that can be used at the clinic immediately (8). The strategy to couple Miro1-based therapy with a Miro1-dependent companion diagnostic tool could significantly improve the success of future clinical trials. This work has formed the foundation of 4 patents with our trainees as co-inventors and a startup company for treating PD.
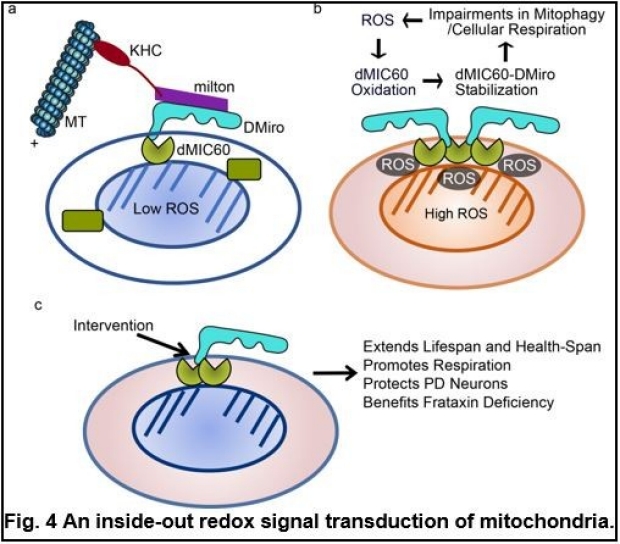
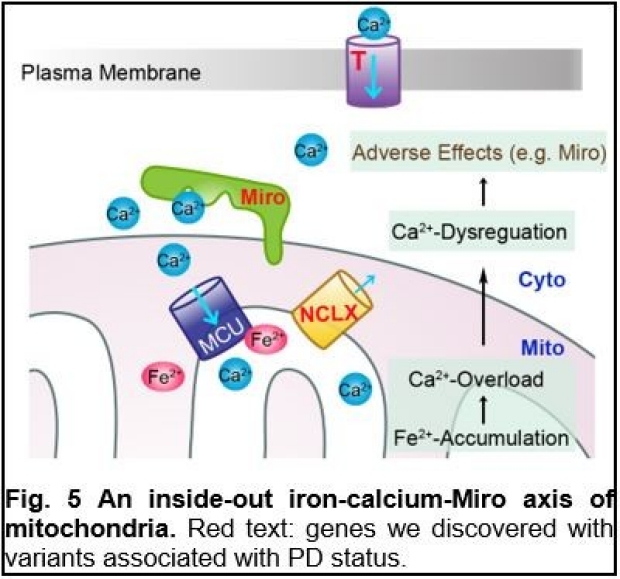
The unifying involvement of Miro in different types of PD suggests complex upstream signals of Miro. Driven by this question, we have found novel mechanisms for inside-out signal transduction of the mitochondria. The molecular pathways of signal transduction from the mitochondrial matrix to the cytosol are poorly defined. We have found that ROS generated during oxidative-stressed conditions such as aging, PD, and Friedreich’s Ataxia (FA), oxidize MIC60, an inner mitochondrial membrane (IMM) protein, leading to oxidative conformational changes of the MIC60 complex. This causes the physical association of MIC60 with the OMM protein Miro, ultimately eliciting cellular responses that delay mitophagy, impair cellular respiration, and cause oxidative stress. Blocking the MIC60-Miro interaction or reducing either protein, genetically or pharmacologically, extends lifespan and health-span of healthy fruit flies, and benefits multiple models of PD and FA (Fig. 4) (6). Our discovery provides a molecular basis for common treatment strategies against oxidative stress. In addition to ROS, recently we have found that intramitochondrial iron can influence cytosolic Ca2+ signals. Elevation of matrix Fe2+-levels causes Ca2+-overflow into the mitochondria, through an interaction of Fe2+ with the mitochondrial calcium uniporter (MCU) complex, the Ca2+-import channel in IMM, and resultant MCU oligomerization. Mitochondrial Ca2+-overload causes cellular Ca2+-dysregulation, which could be sensed by Miro, the OMM protein with the Ca2+-binding domains facing the cytosol (Fig. 5). Thus, this mechanism allows instantaneous sensing of Fe2+ inside the mitochondria to influence cytosolic Ca2+ across the mitochondrial double membranes, and provides a paradigm for how one ion (iron) could affect another ion (calcium) on a spatial and temporal scale. Pathologically, this mechanism is activated in human neuron models and postmortem brains of patients with PD and can be targeted by multiple ways for alleviating neuron loss (8).
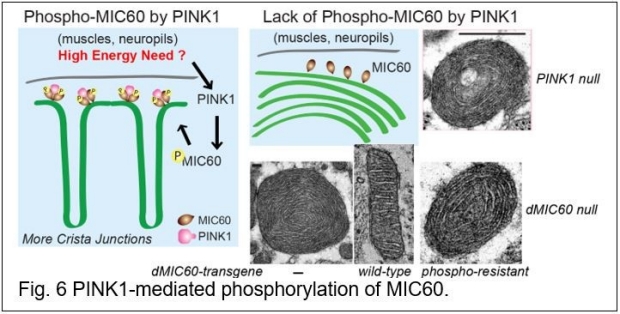
We have also been searching for PINK1’s substrates in addition to Miro. By employing phospho-proteomic approaches in flies, we have discovered multiple novel substrates of PINK1 inside the healthy mitochondria. One is the inner membrane protein, MIC60/mitofilin, and the PINK1-MIC60 pathway maintains remodeling of crista junctions, the complex I activity, and dopaminergic neuronal survival in vivo. Importantly, by combining human genetics and unbiased functional screens in flies, we have identified human MIC60 mutations that are highly damaging in vivo and may increase the risk of PD in humans. Our finding represents an example of how diseases may arise when the fundamental principles go awry (Fig. 6) (9). Another substrate regulates fatty acid beta-oxidation (MCAD). We have found that the restoration of phosphorylation of MCAD significantly rescues PINK1 null’s organismal phenotypes. We have further examined the acryl-carnitine and metabolite profiles in flies with or without PINK1-mediated phosphorylation of MCAD and identified surprising and substantial metabolite abnormalities. This work adds a new layer of the current understandings of PINK1’s physiological role in metabolic homeostasis (10).
Our work, together with others’, shows that mitochondria are the center for maintaining neuronal homeostasis, cellular communications, and signal transduction. We are continuing to study the underlying mechanisms. For example, how do mitochondria communicate with other organelles in subdomains of neurons? What signals dictate mitochondria to permanently stay, pause, or move? How is mitochondrial damage cleared or mended at the distal synapses? We will address these questions by studying subcellular domain-specific mechanisms, as well as system-level research on the coordination of neuronal mitochondria with other organelles and neuron-glia interactions, to reveal the impact of mitochondria on the nervous system at both the molecular and organismal level.

Reference:
1. Wang X, Schwarz TL. The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136(1):163-74. doi: 10.1016/j.cell.2008.11.046. PubMed PMID: 19135897; PMCID: 2768392.
2. Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147(4):893-906. Epub 2011/11/15. doi: 10.1016/j.cell.2011.10.018. PubMed PMID: 22078885; PMCID: PMC3261796.
3. Hsieh CH, Li L, Vanhauwaert R, Nguyen KT, Davis MD, Bu G, Wszolek ZK, Wang X. Miro1 Marks Parkinson's Disease Subset and Miro1 Reducer Rescues Neuron Loss in Parkinson's Models. Cell metabolism. 2019;30(6):1131-40 e7. Epub 2019/10/01. doi: 10.1016/j.cmet.2019.08.023. PubMed PMID: 31564441; PMCID: PMC6893131.
4. Hsieh CH, Shaltouki A, Gonzalez AE, Bettencourt da Cruz A, Burbulla LF, St Lawrence E, Schule B, Krainc D, Palmer TD, Wang X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson's Disease. Cell stem cell. 2016;19(6):709-24. Epub 2016/09/13. doi: 10.1016/j.stem.2016.08.002. PubMed PMID: 27618216; PMCID: PMC5135570.
5. Shaltouki A, Hsieh CH, Kim MJ, Wang X. Alpha-synuclein delays mitophagy and targeting Miro rescues neuron loss in Parkinson's models. Acta Neuropathol. 2018;136(4):607-20. Epub 2018/06/21. doi: 10.1007/s00401-018-1873-4. PubMed PMID: 29923074; PMCID: PMC6123262.
6. Li L, Conradson DM, Bharat V, Kim MJ, Hsieh CH, Minhas PS, Papakyrikos AM, Durairaj AS, Ludlam A, Andreasson KI, Partridge L, Cianfrocco MA, Wang X. A mitochondrial membrane-bridging machinery mediates signal transduction of intramitochondrial oxidation. Nat Metab. 2021. Epub 2021/09/11. doi: 10.1038/s42255-021-00443-2. PubMed PMID: 34504353.
7. Nguyen D, Bharat V, Conradson DM, Nandakishore P, Wang X. Miro1 Impairment in a Parkinson's At-Risk Cohort. Front Mol Neurosci. 2021;14:734273. Epub 2021/08/27. doi: 10.3389/fnmol.2021.734273. PubMed PMID: 34434090; PMCID: PMC8381147.
8. Vinita Bharat RV, Li Li, Colin M. Muir, Aarooran Sivakumaran Durairaj, Sujyoti Chandra, Yann Le Guen, Pawan Nandakishore, Chung-Han Hsieh, Stefano E. Rensi, Russ B. Altman, Michael D. Greicius, Liang Feng, Xinnan Wang. An Iron-Calcium-Miro Axis Influences Parkinson’s Risk and Neurodegeneration. In: University S, editor. BioRxiv2022.
9. Pei-I Tsai, Chin-Hsien Lin, Chung-Han Hsieh, Amanda M Papakyrikos, Min Joo Kim, Valerio Napolioni, Carmen Schoor, Julien Couthouis, Ruey-Meei Wu, Zbigniew K. Wszolek, Dominic Winter, Michael D. Greicius, Owen A. Ross, and Xinnan Wang. (2018) PINK1 Phosphorylates MIC60/Mitofilin to Control Structural Plasticity of Mitochondrial Crista Junctions. Molecular Cell 69(5):744-756. PMID: 29456190.
10. Meredith M. Course, Anna I. Scott, Carmen Schoor, Chung-Han Hsieh, Amanda M. Papakyrikos, Dominic Winter, Tina M. Cowan, and Xinnan Wang (2018) Phosphorylation of MCAD selectively rescues PINK1 deficiencies in behavior and metabolism. Molecular Biology of the Cell doi:10.1091/mbc.E18-03-0155.